What is Charcot-Marie-Tooth?
|
Charcot-Marie-Tooth is the most common genetic disorder involving the peripheral nervous system, affecting an estimated 126,000 people in the United States and 2.6 million worldwide.
Charcot-Marie-Tooth is a disease involving the demyelination of nerves in the peripheral nervous system. This demyelination results in the atrophy of muscles in the legs, hands, and feet, as well as the loss of sensation in these areas. This often results in symptoms such as the presence of stork legs, hammer toes and high arches of the feet. The disease is progressive and currently has no cure, although it is not considered to be life-threatening. Onset usually occurs in adolescence or early adulthood and Charcot-Marie-Tooth usually live normal lifespans and the condition can be helped via supportive care. |
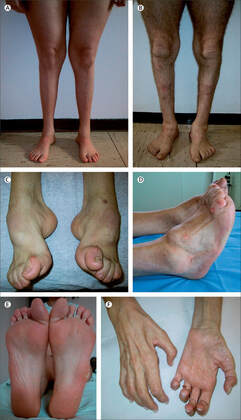
Diagnosis, natural history, and management of Charcot–Marie–Tooth disease
Pareyson, Davide et al.
The Lancet Neurology, Volume 8, Issue 7, 654 - 667
|
PMP22 is mutated in Charcot-Marie-Tooth
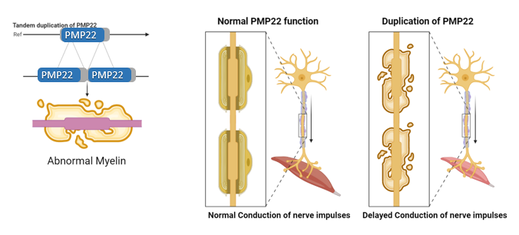
|
The PMP22 gene is duplicated in the case of Charcot-Marie-Tooth Type 1A. This duplication results in a toxic overexpression of the PMP22 gene product, resulting in the demyelination and other symptoms seen. Myelin plays an imperative role in the sending of messages to and from the brain and duplication of PMP22 causes atrophy of the muscles in the hands and feet through the delayed conduction of nerve impulses as a result of the demyelination.
Specifically, PMP22 can be shown to function in the myelination of Schwann cells and specifically have Nucleoside Triphosphatase Activity. |
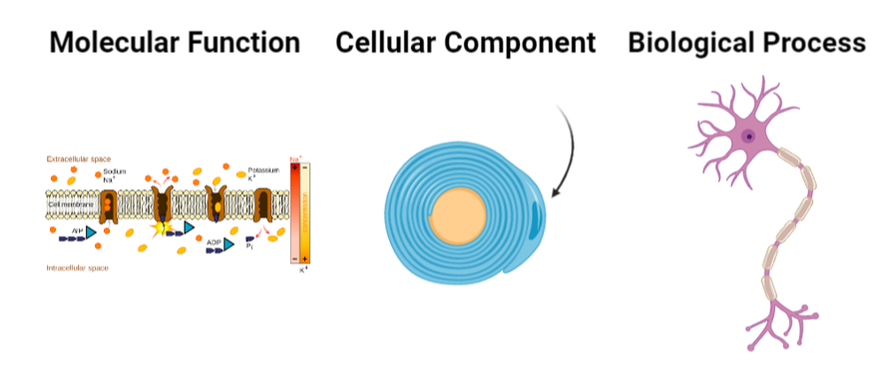
|
Nuceloside Triphosphatase Acivity
|
Schwann Cells
|
Myelination
|
PMP22 can also be shown to be highly conserved across other species. Specifically, it is well conserved among species with extensive peripheral nervous systems such as chimpanzees, macaques, dogs, mice and rats. This makes sense as PMP22 has been implicated in having a role in myelination. In particular, PMP22 can be seen to be well conserved in mice, a excellent model organism for studying the peripheral nervous system. This is evident through their relative closeness in the phylogenetic tree below and evidence that the protein domain is well conserved.
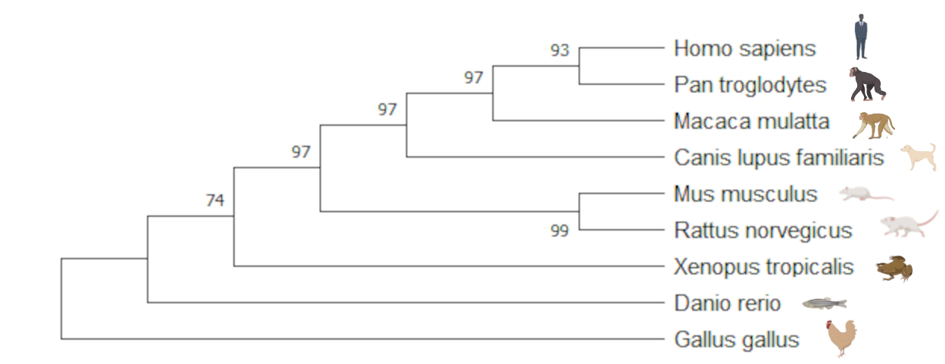
Phylogenetic Tree of PMP22
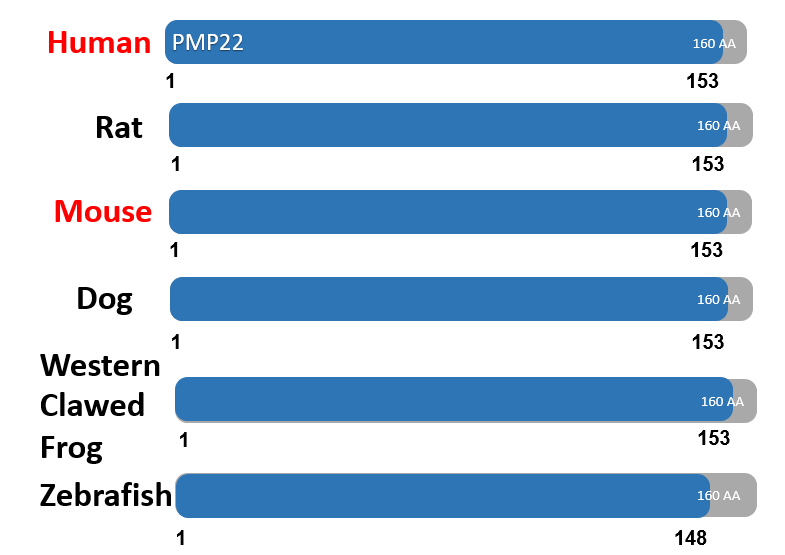
The PMP22 protein domain is well conserved in model organisms
Looking at the already known protein interactions of PMP22 shows that it interacts heavily with proteins involved in myelination. These interactions can help shine a light on the possible function of PMP22 in the formation of myelin sheath. In addition to this, the knowledge of these already existing protein interactions can help in the discovery of changed protein interactions in individuals with PMP22 mutations, and the presence of novel protein interactions that have not yet been discovered. Both of these future areas of research will help the arrival of a potential treatment for Charcot-Marie-Tooth
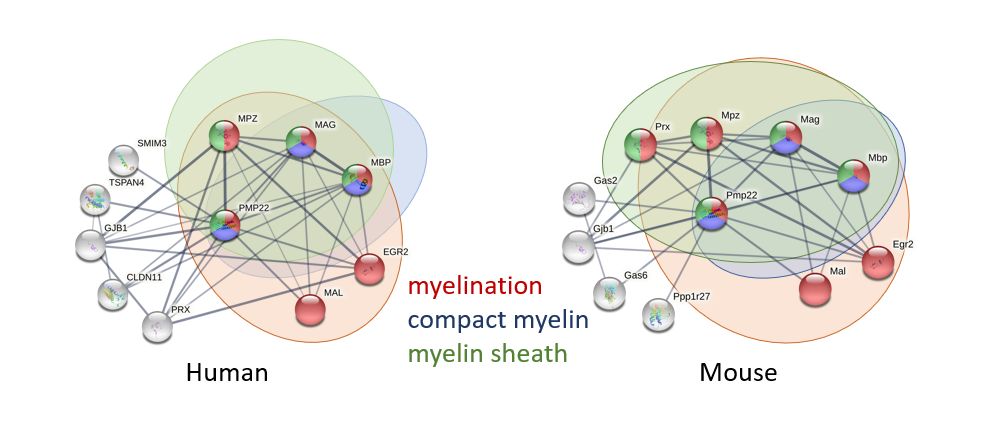
Mice as a model organism
Rosso, G., Cal, K., Canclini, L., Damián, J. P., Ruiz, P., Rodríguez, H., Sotelo, J. R., Vazquez, C., & Kun, A. (2010). Early phenotypical diagnoses in Trembler-J mice model
|
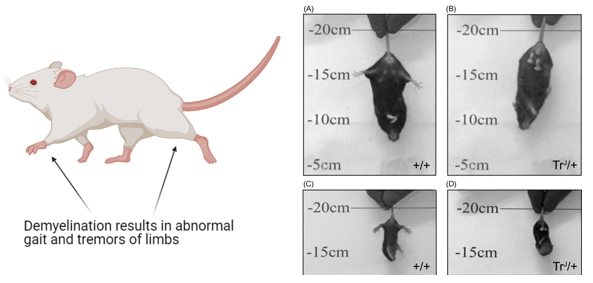
Low, P. A., & McLeod, J. G. (1975). Hereditary demyelinating neuropathy in the Trembler mouse
|
Mice act as excellent model organisms for studying the peripheral nervous system due to the ease of assaying myelin formation. Mice have complex peripheral nervous systems similar to humans and as such, PMP22 mutations cause similar phenotypes in mice as they do in humans. An example of this can be seen above in which a mouse on the right with PMP22 mutations can be seen to show abnormal limb positioning when compared to the wildtype mouse on the right.
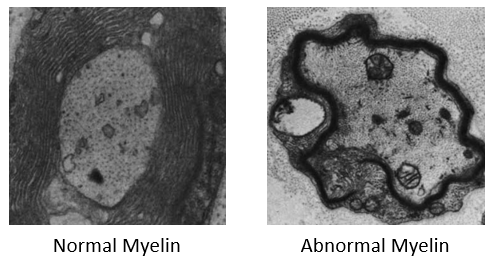
In addition to this, the sciatic nerve of these mice can easily be harvested an viewed under the microscope, producing images seen on the right. This ability to observe the myelinating state of these nerve cells is an excellent way to study PMP22s role in myelination.
In addition to this, the sciatic nerve of these mice can easily be harvested an viewed under the microscope, producing images seen on the right. This ability to observe the myelinating state of these nerve cells is an excellent way to study PMP22s role in myelination.
Gap in knowledge
|
It is currently unknown what the exact role of PMP22 is in the formation of myelin.
Thus, I hypothesize that the glycoprotein PMP22 in the peripheral nervous system plays a role in myelination through protein-protein interactions with other proteins |
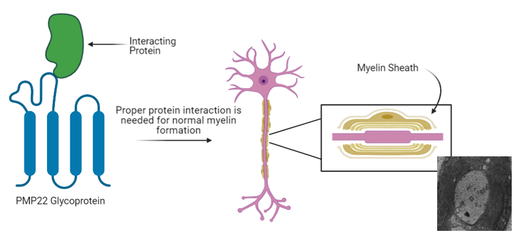
|
Exploration of this knowledge gap
Aim 1: Identify conserved amino acids in PMP22 necessary for the regulation of myelination.

|
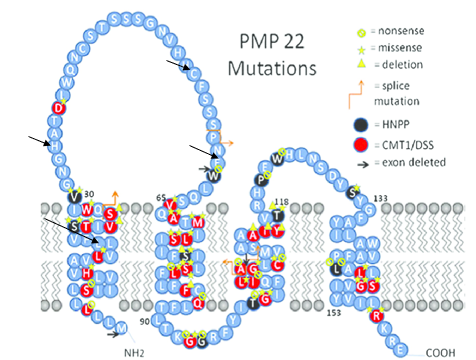
|
Amino acids 34, 53, and 60 are located in the extracellular loop portion of the PMP22 protein and are likely to be involved in protein interactions. Therefor, mutations in these regions involved in protein interactions may result in demyelination. Thus, these proteins will be mutated via a CRISPR-Cas9 system in order to produce mutants with the demyelinating phenotype. In addition to this, amino acid 18, which is in the intermembrane space of the protein will also be mutated as a control. This will act as a control as due to being in the intermembrane space, it should not impact protein interactions and therefore should not result in demyelination. These mice will have their sciatic nerve harvested and the myelinating status of the cells will be observed. Both the wildtype and those mutants with the demyelination will have the sciatic nerve cells saved for the next aims.
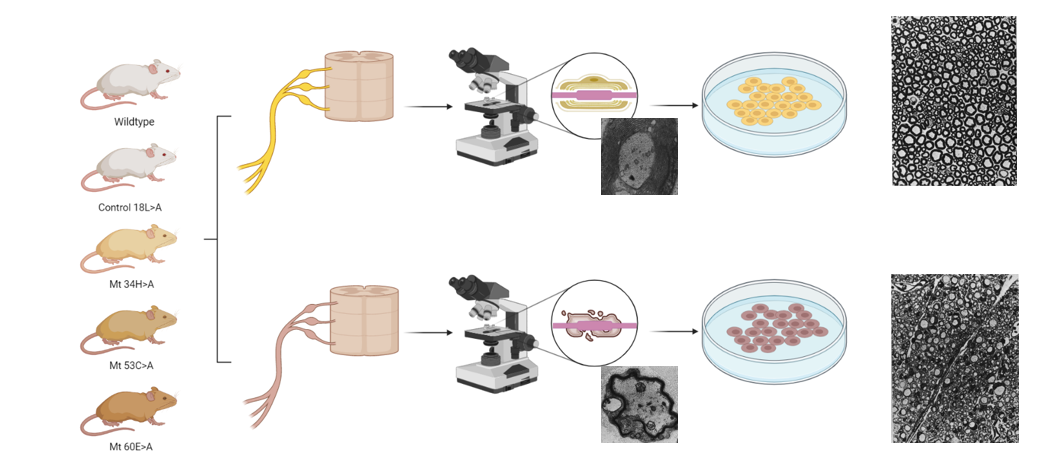
Rationale - Identifying amino acids that impact myelination can give insight into how mutations of PMP22 may result in demyelination.
I hypothesize that mice with mutations in conserved PMP22 amino acids in these extracellular loop regions will show demyelination in the peripheral nervous system
I hypothesize that mice with mutations in conserved PMP22 amino acids in these extracellular loop regions will show demyelination in the peripheral nervous system
Aim 2: Identify genes that are differentially expressed in in WT and PMP22 mutant myelinating Schwann cells.
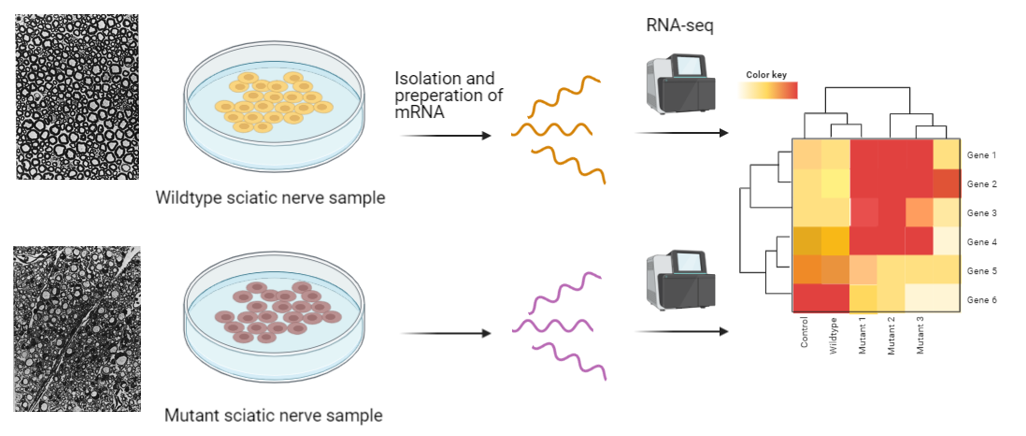
|
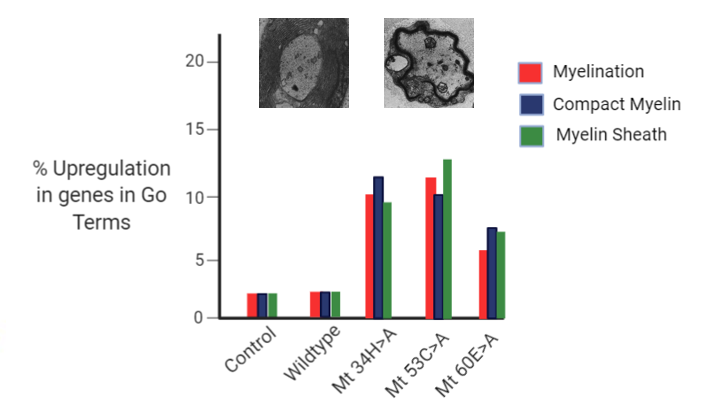
|
The sciatic nerve samples taken from the demyelinating mutant and the wildtype will have RNA isolated and RNA-seq will be ran. This gene expression data seen in the heat map will be sorted via Gene Ontology labels. Those genes that are differentially expressed in the wildtype vs the mutant mice will be grouped into the Gene Ontology groups - myelination, compact myelin, and myelin sheath. Then, the percent gene expression will be compared between the wildtype and the mutants in order to find the percent upregulation of these genes involved in myelination in the mutants.
Rationale - Identification of these differentially transcribed genes will aid in determining what genes and pathways PMP22 interacts with in myelination.
Thus, I hypothesize that Mice with PMP22 mutations will show abnormal levels of gene expression in pathways related to myelination. This will be due to the the altered protein interactions in the mutant mice altering transcription levels of genes related to myelination
This data can be confirmed by doing RT-qPCR with the same isolated RNA from both the wildtype and the mutant mice, which when compared to the data gathered from RNA-seq, will validate any results gained.
Rationale - Identification of these differentially transcribed genes will aid in determining what genes and pathways PMP22 interacts with in myelination.
Thus, I hypothesize that Mice with PMP22 mutations will show abnormal levels of gene expression in pathways related to myelination. This will be due to the the altered protein interactions in the mutant mice altering transcription levels of genes related to myelination
This data can be confirmed by doing RT-qPCR with the same isolated RNA from both the wildtype and the mutant mice, which when compared to the data gathered from RNA-seq, will validate any results gained.
Aim 3: Identify novel proteins important for myelination.
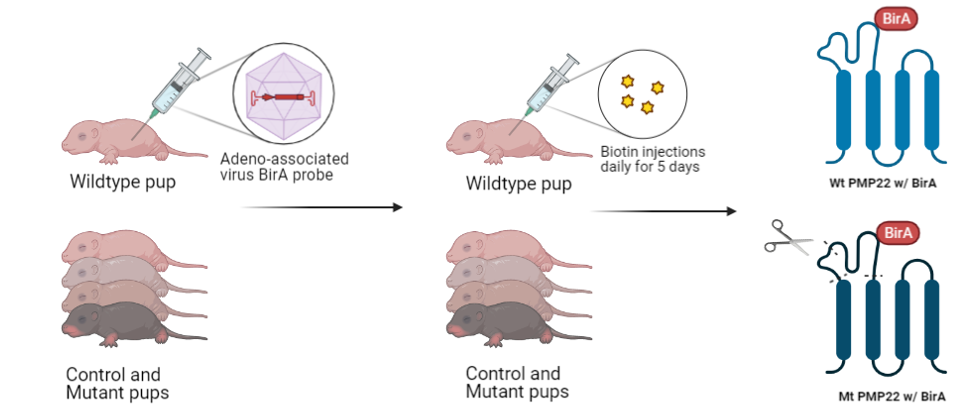
For aim three, the same mutant, control, and wildtype mice created using CRISPR-Cas9 will be injected with an adeno-associated virus with a BirA probe. This BirA probe is a part of the BioID system for detecting proximity labeled protein interactions. This process will create PMP22 proteins with the BirA probe attached in both the wildtype and the mutant. Next, shown in the process below, Biotin will be injected into the mice pups resulting in the labeling of proteins closely associated with PMP22. By harvesting these proteins, isolating them, and running them through mass spectrometry, the identity of these novel proteins interacting with PMP22 can be found.
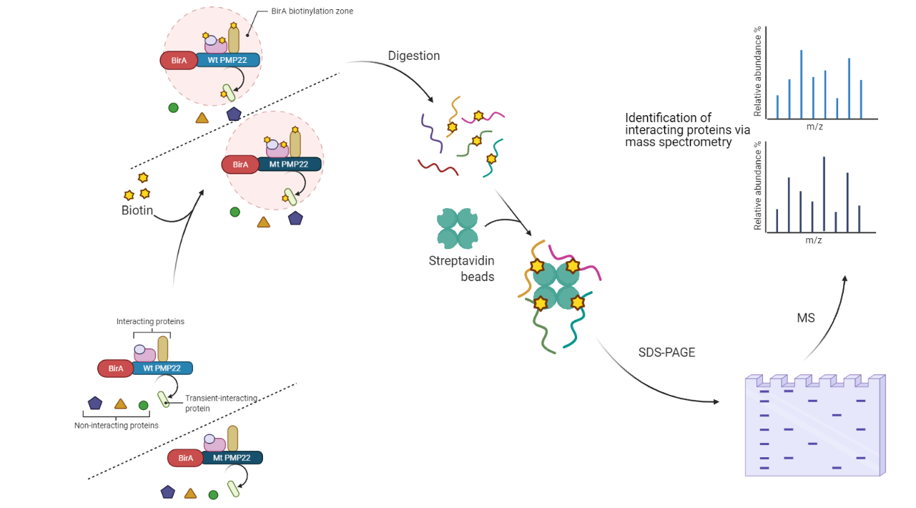
Rationale - Discovery of new protein interactions that are impacted by PMP22 mutation can help enlighten the mechanisms by which PMP22 impacts myelination.
Thus, I hypothesize that analysis of M/Z data from BioID will elucidate new protein interactions and differences between wildtype & mutant PMP22 ability to interact with other proteins.
Thus, I hypothesize that analysis of M/Z data from BioID will elucidate new protein interactions and differences between wildtype & mutant PMP22 ability to interact with other proteins.
The future directions of research
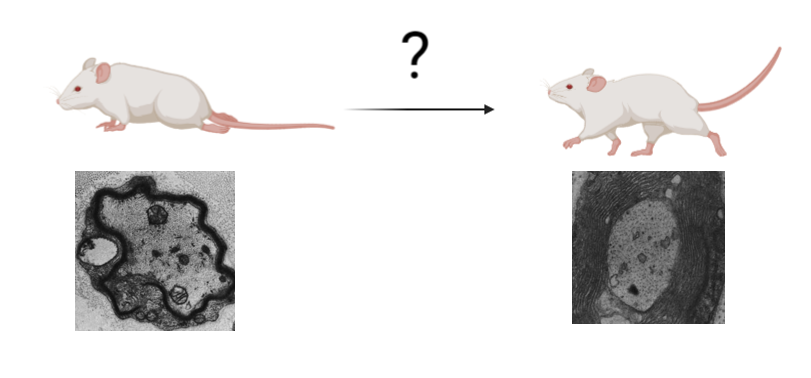
For the future directions of research involving PMP22 and the peripheral nervous system, I am interested in using this new knowledge of protein interactions and their function in PMP22 and myelination for the rescuing of the demyelinating phenotype. Specifically I would likely to research how exactly we can use this knowledge of protein interactions to create a long term treatment for those affected by Charcot-Marie-Tooth and other demyelinating diseases of the peripheral nervous system. Discovery of mall molecules that fix these protein interactions could also be a possibility, and therefore a small molecule screen should be done in the presence of these mutations in hopes of rescuing the phenotype.
Presentations
| nguyen20april2021final.pdf |
| |||
| nguyen4march2021.pdf |
References
Biorender used to create images
Li, J., Parker, B., Martyn, C., Natarajan, C., & Guo, J. (2013). The PMP22 Gene and Its Related Diseases. Molecular Neurobiology, 47(2), 673–698. https://doi.org/10.1007/s12035-012-8370-x
Robertson, A. M., Perea, J., McGuigan, A., King, R. H. M., Muddle, J. R., Gabreels-Festen, A. A., Thomas, P. K., & Huxley, C. (2002). Comparison of a new pmp22 transgenic mouse line with other mouse models and human patients with CMT1A*. Journal of Anatomy, 200(4), 377–390. https://doi.org/10.1046/j.1469-7580.2002.00039.x
Inoue, K. (2001). The 1.4-Mb CMT1A Duplication/HNPP Deletion Genomic Region Reveals Unique Genome Architectural Features and Provides Insights into the Recent Evolution of New Genes. Genome Research, 11(6), 1018–1033. https://doi.org/10.1101/gr.180401
Jetten, A. M. (n.d.-b). The Peripheral Myelin Protein 22 and Epithelial Membrane Protein Family. 33.
Knapp, P. E. (1996). Proteolipid Protein: Is It More than Just a Structural Component of Myelin? Developmental Neuroscience, 18(4), 297–308. https://doi.org/10.1159/000111420
Low, P. A., & McLeod, J. G. (1975). Hereditary demyelinating neuropathy in the Trembler mouse. Journal of the Neurological Sciences, 26(4), 565–574. https://doi.org/10.1016/0022-510X(75)90057-X
Mittendorf, K. F., Marinko, J. T., Hampton, C. M., Ke, Z., Hadziselimovic, A., Schlebach, J. P., Law, C. L., Li, J., Wright, E. R., Sanders, C. R., & Ohi, M. D. (2017). Peripheral myelin protein 22 alters membrane architecture. Science Advances, 3(7), e1700220. https://doi.org/10.1126/sciadv.1700220
Rosso, G., Cal, K., Canclini, L., Damián, J. P., Ruiz, P., Rodríguez, H., Sotelo, J. R., Vazquez, C., & Kun, A. (2010). Early phenotypical diagnoses in Trembler-J mice model. Journal of Neuroscience Methods, 190(1), 14–19. https://doi.org/10.1016/j.jneumeth.2010.04.010
Watila, M. M., & Balarabe, S. A. (2015). Molecular and clinical features of inherited neuropathies due to PMP22 duplication. Journal of the Neurological Sciences, 355(1–2), 18–24. https://doi.org/10.1016/j.jns.2015.05.037
Li, J., Parker, B., Martyn, C., Natarajan, C., & Guo, J. (2013). The PMP22 Gene and Its Related Diseases. Molecular Neurobiology, 47(2), 673–698. https://doi.org/10.1007/s12035-012-8370-x
Robertson, A. M., Perea, J., McGuigan, A., King, R. H. M., Muddle, J. R., Gabreels-Festen, A. A., Thomas, P. K., & Huxley, C. (2002). Comparison of a new pmp22 transgenic mouse line with other mouse models and human patients with CMT1A*. Journal of Anatomy, 200(4), 377–390. https://doi.org/10.1046/j.1469-7580.2002.00039.x
Inoue, K. (2001). The 1.4-Mb CMT1A Duplication/HNPP Deletion Genomic Region Reveals Unique Genome Architectural Features and Provides Insights into the Recent Evolution of New Genes. Genome Research, 11(6), 1018–1033. https://doi.org/10.1101/gr.180401
Jetten, A. M. (n.d.-b). The Peripheral Myelin Protein 22 and Epithelial Membrane Protein Family. 33.
Knapp, P. E. (1996). Proteolipid Protein: Is It More than Just a Structural Component of Myelin? Developmental Neuroscience, 18(4), 297–308. https://doi.org/10.1159/000111420
Low, P. A., & McLeod, J. G. (1975). Hereditary demyelinating neuropathy in the Trembler mouse. Journal of the Neurological Sciences, 26(4), 565–574. https://doi.org/10.1016/0022-510X(75)90057-X
Mittendorf, K. F., Marinko, J. T., Hampton, C. M., Ke, Z., Hadziselimovic, A., Schlebach, J. P., Law, C. L., Li, J., Wright, E. R., Sanders, C. R., & Ohi, M. D. (2017). Peripheral myelin protein 22 alters membrane architecture. Science Advances, 3(7), e1700220. https://doi.org/10.1126/sciadv.1700220
Rosso, G., Cal, K., Canclini, L., Damián, J. P., Ruiz, P., Rodríguez, H., Sotelo, J. R., Vazquez, C., & Kun, A. (2010). Early phenotypical diagnoses in Trembler-J mice model. Journal of Neuroscience Methods, 190(1), 14–19. https://doi.org/10.1016/j.jneumeth.2010.04.010
Watila, M. M., & Balarabe, S. A. (2015). Molecular and clinical features of inherited neuropathies due to PMP22 duplication. Journal of the Neurological Sciences, 355(1–2), 18–24. https://doi.org/10.1016/j.jns.2015.05.037
